
In the last decade there has been a tremendously growing effort aimed at integrating organic materials into electronic devices. This is driven by the opportunity to fabricate devices by cheap chemical methods that can be implemented on flexible substrates, thus enabling disposable, flexible and wearable electronics. In addition there is also a growing activity in studying spin phenomena in organic materials. Organics are characterized by weak spin-orbit and hyperfine coupling so that the typical spin diffusion times are long. However, their mobility is way inferior to that of inorganic semiconductors, so that the spin-relaxation lengths remain short. Our research program aims at constructing a fully ab initio theory of charge and spin-transport in single-crystal molecular devices. Here are a few on-going projects in this area:
Multi-scale theory of charge and spin transport in organic single crystals
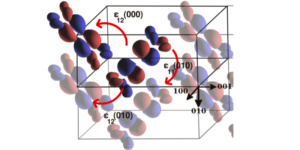 The main feature characterizing small-molecules organic-crystal semiconductors (e.g. rubrene, durene, pentacene, ….) is the fact that the typical bandwidth (about 100meV) is of the same order of magnitude than the energy associated to the electron-phonon coupling. This means that phonons cannot be treated as a perturbation, but need to be built in explicitly in the theory. We have developed a rigorous protocol to evaluate spin and charge transport properties of organic semiconductors. This is a multiscale method that allows one to determine, entirely from first principles, the charge and spin mobility as a function of temperature for any given organic crystal. Our method consists in four main steps. First we calculate the electronic structure of the given organic crystal by mean of density functional theory (DFT) and by taking particular care for the precise determination of the crystal structure. Such step requires the use of advanced van der Waals DFT functionals. Next we construct an effective tight-binding Hamiltonian by projecting the calculated band structures onto a maximally localized Wannier function basis set. In the case of organic materials these coincide with the natural molecular orbital of the molecules. Then we compute the crystal phonon spectrum and evaluate, still by using the Wannier construction, the electron-phonon matrix elements. Finally, the mobility is computed either with mean-field small-polaron theory or with the Kubo’s response theory and Monte Carlo simulations.
The main feature characterizing small-molecules organic-crystal semiconductors (e.g. rubrene, durene, pentacene, ….) is the fact that the typical bandwidth (about 100meV) is of the same order of magnitude than the energy associated to the electron-phonon coupling. This means that phonons cannot be treated as a perturbation, but need to be built in explicitly in the theory. We have developed a rigorous protocol to evaluate spin and charge transport properties of organic semiconductors. This is a multiscale method that allows one to determine, entirely from first principles, the charge and spin mobility as a function of temperature for any given organic crystal. Our method consists in four main steps. First we calculate the electronic structure of the given organic crystal by mean of density functional theory (DFT) and by taking particular care for the precise determination of the crystal structure. Such step requires the use of advanced van der Waals DFT functionals. Next we construct an effective tight-binding Hamiltonian by projecting the calculated band structures onto a maximally localized Wannier function basis set. In the case of organic materials these coincide with the natural molecular orbital of the molecules. Then we compute the crystal phonon spectrum and evaluate, still by using the Wannier construction, the electron-phonon matrix elements. Finally, the mobility is computed either with mean-field small-polaron theory or with the Kubo’s response theory and Monte Carlo simulations.
References:
[2] C. Motta and S. Sanvito. Charge transport properties of durene crystals from first principles. J. Chem. Theo. Comp. 10, 4624 (2014). [1] Sandip Bhattacharya, Akinlolu Akande, and Stefano Sanvito. Spin transport properties of triarylamine-based nanowires. Chem. Comm. 50, 6626 (2014).
Constrained DFT for level alignment at an organic/inorganic interface
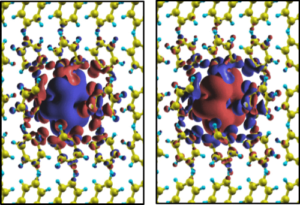 Constrained density functional theory (CDFT) allows one to find the ground state of an electronic system under a given constrain. For instance one can calculate the total energy of a system under the condition that the electronic charge density distributes according to a desired spatial configuration. This becomes particularly useful when there is the need to evaluate quantities associated to the same physical system under different electronic configurations, for instance when describing the charge transfer process between an inorganic surface and a molecule, or when describing the hopping of charge within a molecular crystal. The typical calculation evaluates such intrinsically many-body quantities as total energy differences of configurations implementing different constrains. For instance the charge transfer energy of an electron from a surface to a molecule can be obtained as the total energy difference between the local charge-neutral configuration and that where an electron is taken from the surface and hold on the molecule. The benefit of the method is that calculations are done at the level of local functionals, namely they are computationally cheap, and that several pathological errors associated to DFT are drastically reduced by cancellation when taking the energy differences. The result is a method that, when applicable, returns accuracies at the same level of those of complex many-body techniques (e.g. the GW scheme).
Constrained density functional theory (CDFT) allows one to find the ground state of an electronic system under a given constrain. For instance one can calculate the total energy of a system under the condition that the electronic charge density distributes according to a desired spatial configuration. This becomes particularly useful when there is the need to evaluate quantities associated to the same physical system under different electronic configurations, for instance when describing the charge transfer process between an inorganic surface and a molecule, or when describing the hopping of charge within a molecular crystal. The typical calculation evaluates such intrinsically many-body quantities as total energy differences of configurations implementing different constrains. For instance the charge transfer energy of an electron from a surface to a molecule can be obtained as the total energy difference between the local charge-neutral configuration and that where an electron is taken from the surface and hold on the molecule. The benefit of the method is that calculations are done at the level of local functionals, namely they are computationally cheap, and that several pathological errors associated to DFT are drastically reduced by cancellation when taking the energy differences. The result is a method that, when applicable, returns accuracies at the same level of those of complex many-body techniques (e.g. the GW scheme).
References:
[3] A. Droghetti, I. Rungger, C.D. Pemmaraju, and S. Sanvito. Fundamental gap of molecular crystals via constrained Density Functional Theory. Phys. Rev. B 93, 195208 (2016).
Spin-phonon coupling in organic materials
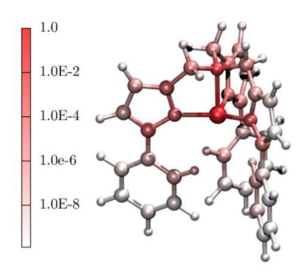 The use of single molecule magnets in mainstream electronics requires their magnetic moment to be stable over long times. One can achieve such a goal by designing compounds with spin-reversal barriers exceeding room temperature, namely with large uniaxial anisotropies. Such strategy, however, has been defeated by several recent experiments demonstrating under-barrier relaxation at high temperature, a behaviour today unexplained. Recently we have proposed spin–phonon coupling to be responsible for such anomaly. With a combination of electronic structure theory and master equations we have shown that, in the presence of phonon dissipation, the relevant energy scale for the spin relaxation is given by the lower-lying phonon modes interacting with the local spins. These open a channel for spin reversal at energies lower than that set by the magnetic anisotropy, producing fast under-barrier spin relaxation. Our research in this area continues with the inclusion in the theory of different sources of spin-relaxation, namely dipolar molecular interaction and hyperfine fields. Our aim is to generate a materials specific theory of spin relaxation in magnetic systems, which can applied to localized and itinerant magnets on the same footing.
The use of single molecule magnets in mainstream electronics requires their magnetic moment to be stable over long times. One can achieve such a goal by designing compounds with spin-reversal barriers exceeding room temperature, namely with large uniaxial anisotropies. Such strategy, however, has been defeated by several recent experiments demonstrating under-barrier relaxation at high temperature, a behaviour today unexplained. Recently we have proposed spin–phonon coupling to be responsible for such anomaly. With a combination of electronic structure theory and master equations we have shown that, in the presence of phonon dissipation, the relevant energy scale for the spin relaxation is given by the lower-lying phonon modes interacting with the local spins. These open a channel for spin reversal at energies lower than that set by the magnetic anisotropy, producing fast under-barrier spin relaxation. Our research in this area continues with the inclusion in the theory of different sources of spin-relaxation, namely dipolar molecular interaction and hyperfine fields. Our aim is to generate a materials specific theory of spin relaxation in magnetic systems, which can applied to localized and itinerant magnets on the same footing.
References:
[1] A. Lunghi, F. Totti, R. Sessoli, and S. Sanvito. The role of anharmonic phonons in under-barrier spin relaxation of single molecule magnets. Nature Communication 8, 14620 (2017).
Monte Carlo methods for charge and spin transport in organics
The calculation of the charge mobility in organic crystals requires treating a many-body problem coupling electrons and phonons. Mean-field theories, such as the small polaron theory, provide a first approximation to the problem and they usually perform well at high temperature. However, upon lowering the temperature effects such as scattering to dynamical disorder start to play a crucial role and the mean-field approach breaks down. An alternative is provided by the Kubo’s formalism implemented within Monte Carlo sampling methods. Within this framework we are developing Monte Carlo algorithms to compute the charge mobility of organic crystals in the presence of electron-phonon coupling with phonons having both Peierls and Holstain form. These have to scale at relatively large sampling cell and should include the possibility to deal with a complex phonon spectrum.